MeDuSA
Introductions
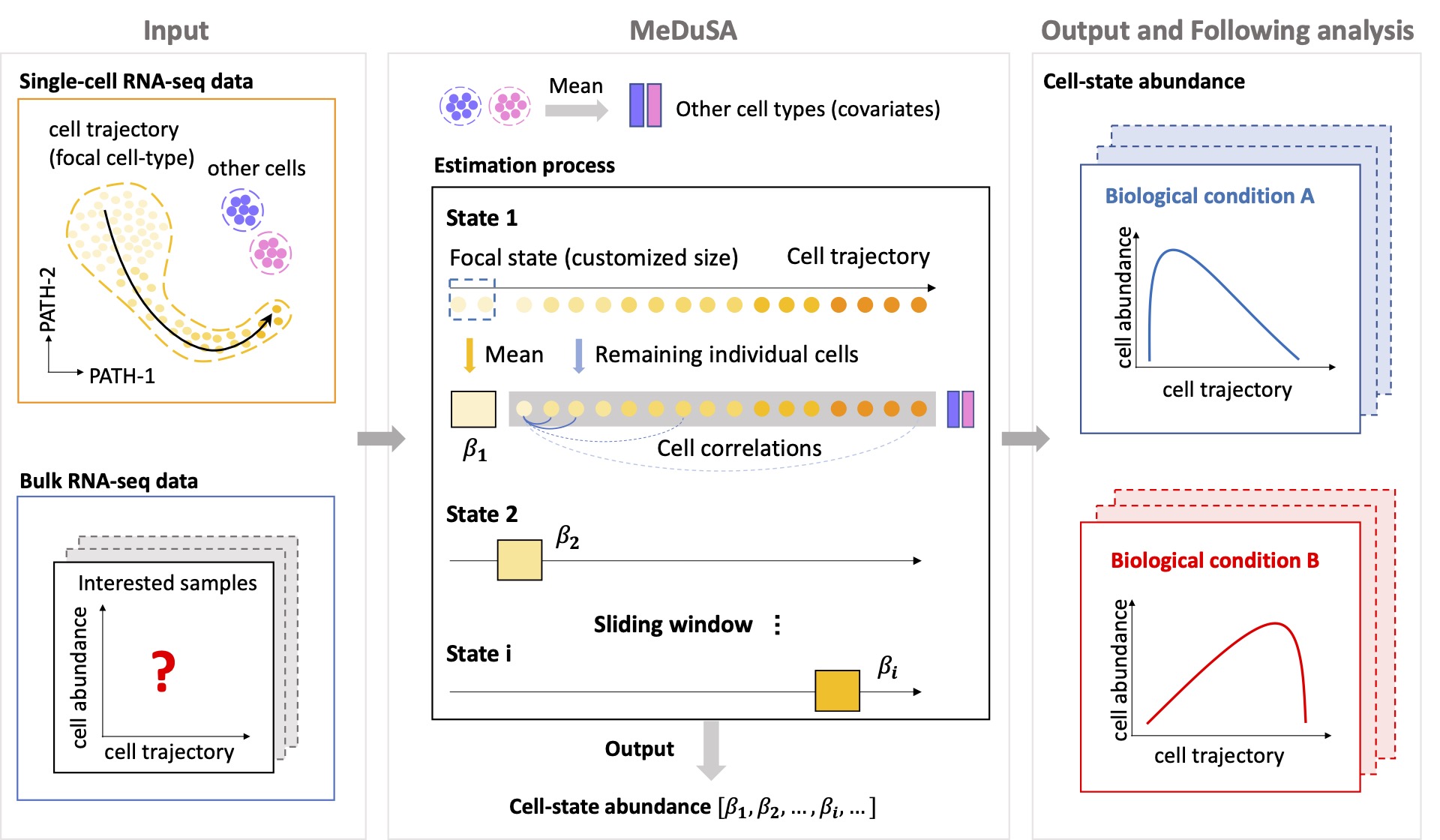
MeDuSA is a cellular deconvolution method that utilizes scRNA-seq data as a reference to estimate cell-state abundance along a one-dimensional trajectory in bulk RNA-seq data. The method employs a linear mixed model (LMM), which fits a cell state in question (either a single cell or the mean of multiple cells) as a fixed effect, while the remaining individual cells of the same cell type as random effects, accounting for the correlations between cells. This innovative model improves deconvolution accuracy by allowing each cell to have a specific weight on bulk gene expression, resulting in a better capture of variance in bulk gene expression. Furthermore, the LMM ameliorates the collinearity problem between cells at the focal state (fitted as a fixed effect) and those at adjacent states (fitted as random effects).
Installations
Extension
MeDuSA now also supports cell-state deconvolution for annotated cell states (cell types). Please check the link below:
https://github.com/LeonSong1995/MeDuSAJ.
MeDuSAJ is more robust for estimating cell state (cell type) abundance for rare cell types, albeit with a slightly increased computational burden. Tutorials can be found in the README of MeDuSAJ.